
近日,我校化工学院刘振教授团队围绕“基于机理认识与人工智能驱动的过渡金属催化剂设计”方向取得系列进展:团队将密度泛函理论(DFT)揭示的速率/选择性控制因素及副产物形成机制,与机器学习模型及可解释性分析相结合,打通了从“机理-参数化-机器学习模型-实验验证”的研究路线,为过渡金属催化体系的高效筛选与定向优化提供了可迁移的方法学支撑。
团队针对CO2与烯烃耦合反应,从反应机理出发,在多构象搜索基础上提取能够反映电子、位阻与几何特征的描述符,从而建立了可用于预测选择性与活性的机器学习模型。研究识别出四类“化学可解释”的关键描述符,催化剂HOMO能级、百分比掩埋体积、八分掩埋体积与咬合角,并指出传统咬合角与位阻之间并非等价,进一步通过“交互项变换策略”解耦多重描述符之间的复杂关系。(Journal of Catalysis, 2026, 454, 116656)
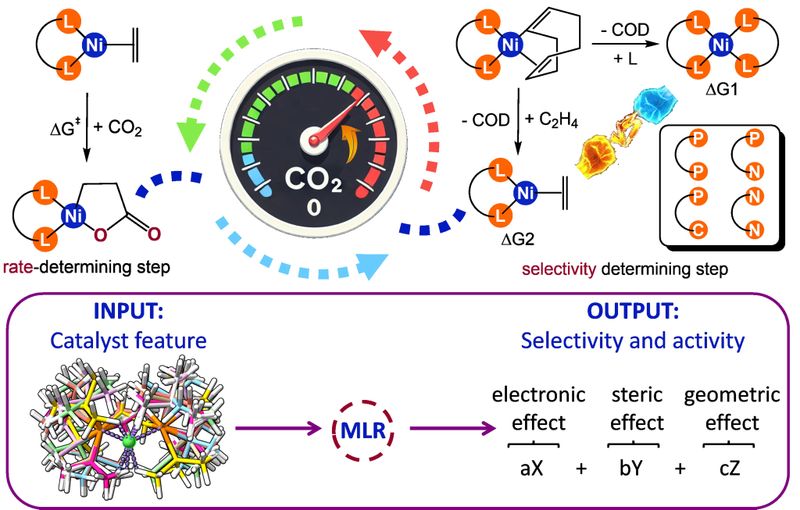
图片说明:围绕速率/选择性决定步骤构建“机理引导”的参数化流程
进一步地,围绕1-己烯与1-辛烯定向合成,团队聚焦Cr/NNN(chromium bis(2-pyridyl)amine)催化体系,构建了包含256个配体的虚拟数据库,并基于DFT计算得到三聚/四聚路径关键过渡态自由能差ΔΔG,结合基于SMILES的结构生成、xTB计算与RDKit结构分析提取分子描述符,建立机器学习预测模型。模型表现出良好的预测能力(R2 = 0.90、MAE = 0.52 kcal/mol),并借助SHAP可解释性分析发现:选择性主要由R1位点的局部位阻控制,而远端电子取代的贡献相对较弱。在模型指导下,团队从数据集中锁定两类新型催化剂,为Cr/NNN体系催化乙烯三聚、四聚制备1-己烯与1-辛烯的可控设计提供了清晰、可迁移的结构规律。(Journal of Catalysis, 2026, 454, 116613)
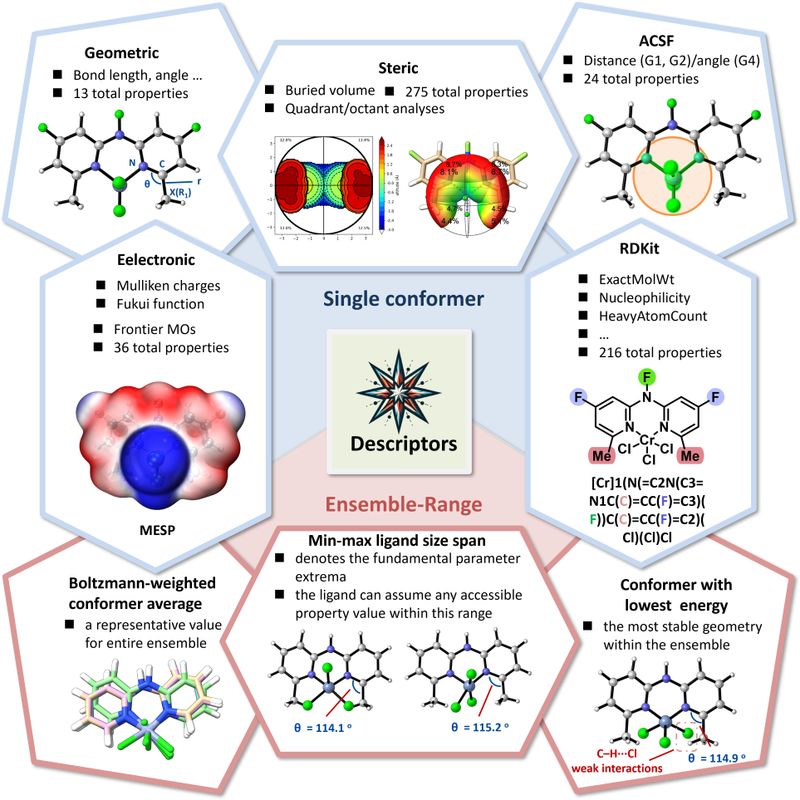
图片说明:基于机理计算的xTB及RDKit描述符集合
针对乙烯三聚、四聚反应中生成的副产物聚乙烯(PE),团队构建了Cr/PNP催化剂数据库、系统提取多尺度分子描述符,并将其与实验测得的PE数据融合训练机器学习模型。研究采用分类-回归双层建模:分类模型可有效区分低/高PE催化剂(accuracy = 0.91),并在工业界关注的低PE区间保持良好召回。对PE < 1 wt%的体系,回归模型可实现较高精度定量预测(R2 = 0.88、MAE = 0.07)。进一步的可解释模型表明:配体本征结构性质决定“低/高PE”分类,而在已属于低PE的配体集合中,优化反应条件(压力、温度、Cr用量、Al/Cr比等)可进一步降低PE含量。(Chemical Science, 2026, https://doi.org/10.1039/D5SC07894F)
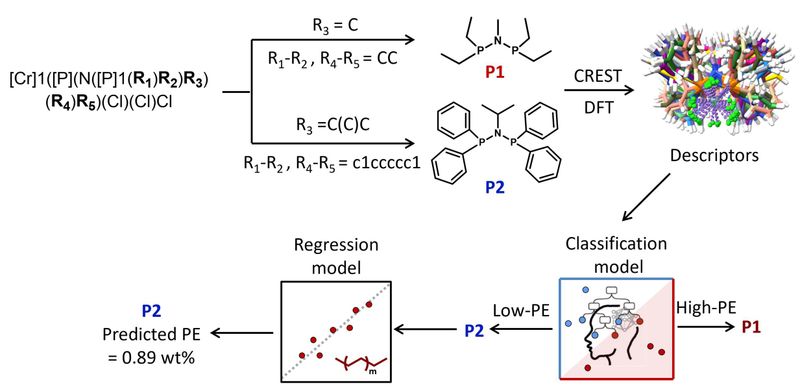
图片说明:用于筛选新型Cr/PNP配体以实现低PE形成的DFT-ML工作流程
论文以华东理工大学为第一通讯单位,我校化工学院博士研究生朱有财同学为第一作者,刘振教授为论文的通讯作者。同时,该研究工作得到了国家自然科学基金面上项目、万华化学的资助。
原文链接:
(1)CO2+C2H4耦合反应催化剂分子设计,Journal of Catalysis 2026: https://doi.org/10.1016/j.jcat.2025.116656
(2)乙烯三聚、四聚催化剂分子设计,Journal of Catalysis 2026: https://doi.org/10.1016/j.jcat.2025.116613
(3)低PE乙烯三聚、四聚催化剂分子设计,Chemical Science 2026: https://doi.org/10.1039/D5SC07894F




