
近日,我校化工学院段学志教授应美国化学协会Accounts of Chemical Research杂志主编Cynthia J. Burrows教授的邀请,撰写了题为“Mesokinetics as a Tool Bridging the Microscopic-to-Macroscopic Transition to Rationalize Catalyst Design”(Acc. Chem. Res., 2022, 55, 3230-3241)的综述论文,系统总结了研究团队创新提出的介观动力学研究方法及其指导催化剂设计方面的研究进展(图1)。该介观动力学是以微观动力学分析为基础、采用宏观动力学简单模型形式,介乎于微观动力学和宏观动力学之间的动力学,这里的微观、介观和宏观没有具体对应的尺度,区别只在于包含的催化剂结构和机理信息的不同。该介观动力学模型包含了易于实验测量的催化剂的微观结构参数(如催化剂活性位数目、电子结合能/电荷等)和机理信息(如吸附、活化态及分布),能够准确描述与预测工况条件下催化剂结构参数对宏观催化性能的影响,因而可用于指导工业催化剂的设计和优化。
图1:介观动力学视角下的催化剂设计
催化反应动力学是工业催化剂和催化反应器设计优化的基础,主要包括催化反应机理和速率方程两部分。在进行反应器设计与开发时,只需要速率方程以计算指定温度和浓度下的反应速率;但在建立速率方程时,为了减少实验工作量,保证反应速率计算的准确性和可靠性,还要求速率方程能正确反映催化剂表面的物理化学过程,这时就要深入研究催化反应机理。但这里所说的催化反应机理,尽管涉及催化剂表面的吸附、脱附、反应基元步骤,也只是将反应温度和浓度/压力与反应速率关联起来,甚至还可与催化剂颗粒内的传递过程关联起来,但并不涉及催化剂的组成、结构和性质,因而难以用于指导催化剂的设计优化。反应动力学是化学反应工程的核心之一,把催化剂的宏观反应条件(温度、压力)和宏观反应结果通过反应机理联系起来。如果对反应机理的研究进一步深入,将催化剂的微尺度结构与物理化学过程通过数学关联起来,就可用于指导催化剂的设计和优化。
论文一方面创新发展了催化反应速率解析的方法,采用原位光谱/动力学分析与多尺度模拟相结合的研究手段,实现基于动力学分析的催化剂活性位与催化描述符辨认。另一方面,提出了“自下而上”的电子效应研究策略(图2),定量关联了“金属-载体表界面相互作用-反应机理-速率决速步的动力学与热力学信息-催化性能”之间的关系,从分子层面阐明了金属-载体间的电子相互作用根源及其对催化动力学行为及反应机理的调控机制,实现了高活性和选择性的催化剂设计开发。
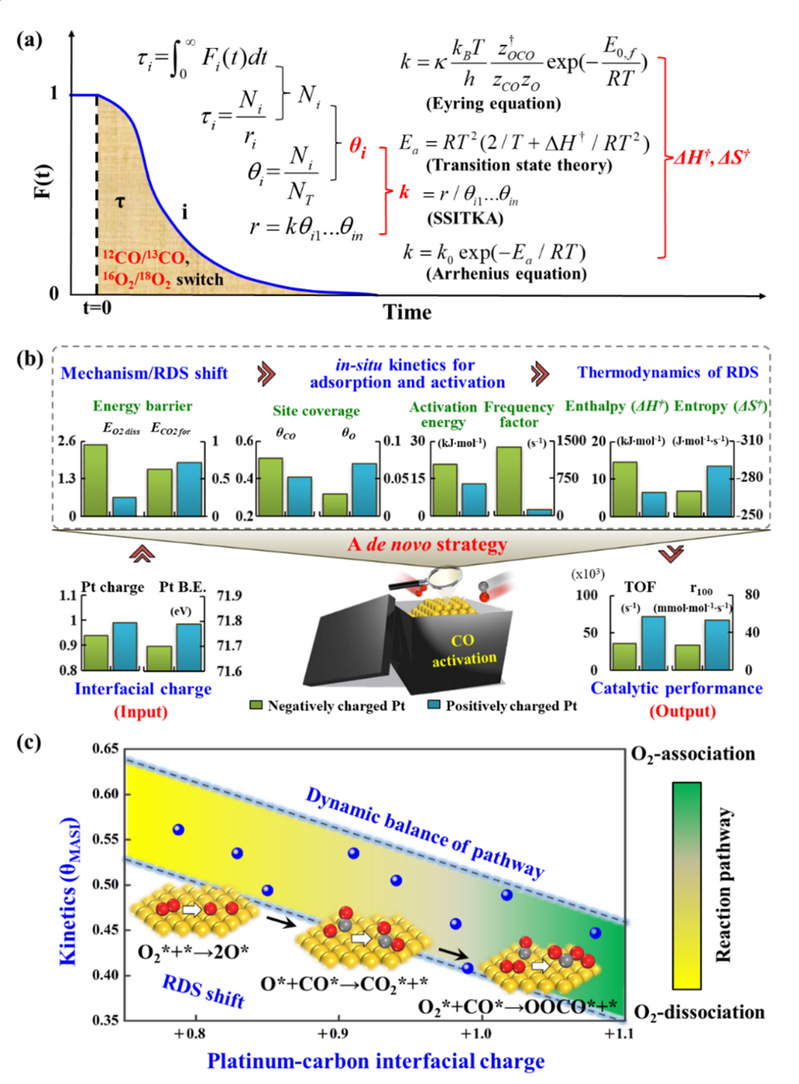
图2:碳载贵金属催化剂“自下而上”的电子效应研究策略
负载型金属催化剂的反应速率(r)可表达为活性位数量(Ni)与活性位活性(TOFi)之积,即r=Ni×TOFi。论文将基于催化剂几何与电子结构的数学表达式导入反应速率方程,从而获得包含金属催化剂活性位数量(N)与活性描述符(d)的介观动力学模型(图3)。该模型能准确定量催化剂微观结构对宏观催化性能的影响,是模型催化剂和真实催化剂之间的一个“桥梁”,也是架起催化剂微观结构和宏观催化性能之间的“桥梁”,促进了催化反应动力学研究向纵深发展,为基于动力学分析的催化剂理性设计提供重要理论基础。
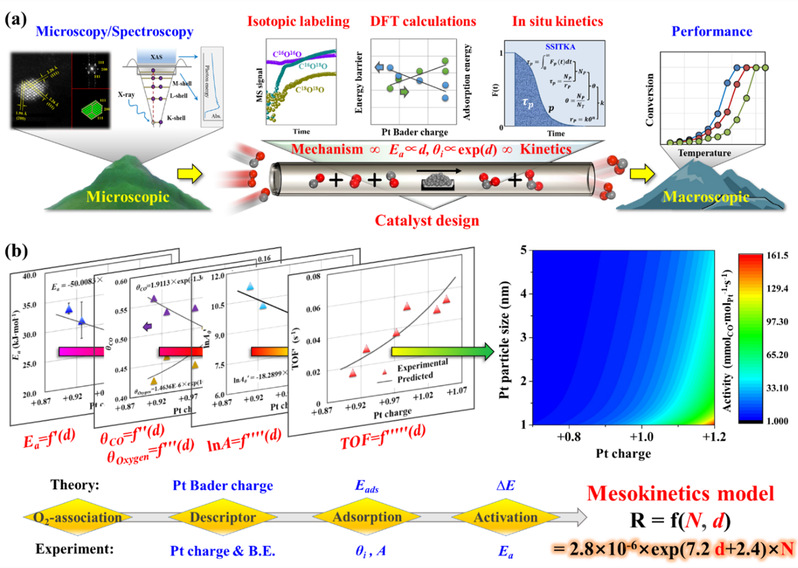
图3:Pt催化CO氧化的介观动力学建模方法
我校陈文尧特聘研究员为论文的第一作者,段学志教授为通讯作者。该研究得到了袁渭康院士、周兴贵教授与挪威科技大学De Chen院士等的悉心指导。此外,该研究工作得到了国家重点研发计划项目、国家自然科学基金重点项目/优秀青年科学基金项目、上海市教委科研创新计划自然科学重大项目、上海市科委科技创新行动计划等项目的支持。
论文链接:https://pubs.acs.org/doi/full/10.1021/acs.accounts.2c00483




