
近日,我校化工学院、化学工程联合国家重点实验室段学志教授、陈文尧博士等从分子层面阐明了金属-载体间的电子相互作用(Electronic Metal–Support Interaction,EMSI)根源及其对催化动力学行为及反应机理的调控机制。相关成果以“Engineering Electronic Platinum-Carbon Support Interaction to Tame Carbon Monoxide Activation”为题发表在Fundamental Research(Fund. Res., DOI: 10.1016/j.fmre.2022.06.012)上。
在本工作中,研究团队基于前期工作基础(Nat. Commun., 2021, 12, 6888; Angew. Chem. Int. Ed., 2022, 61, e202200190),以炭载体表面化学性质对负载金属电子性质的调控为出发点、炭载Pt催化CO氧化为研究体系,以反应动力学为研究主线,采用原位光谱/动力学研究与多尺度模拟相结合的研究方法,定量揭示了金属-炭载体间的电子相互作用对反应动力学行为与机理的影响机制。采用球差电镜与扩展X射线吸收精细结构分析以及X射线光电子能谱与X射线近边吸收光谱分别对Pt催化剂的几何与电子结构进行了解析,在排除催化剂Pt颗粒形貌、粒径、分布等影响下,获得了系列不同电子相互作用(铂-碳界面电荷分布)的催化剂(图1)。在此基础上,通过同位素示踪技术与基于反应力场的分子动力学模拟(ReaxFF-MD)研究了铂-炭载体电子相互作用对反应机理的影响;结合稳态同位素瞬态分析(SSITKA)与DFT理论计算考察了铂-炭载体电子相互作用对反应动力学行为(表面吸附/活化态、关键物种覆盖度、速率决速步等)的影响。
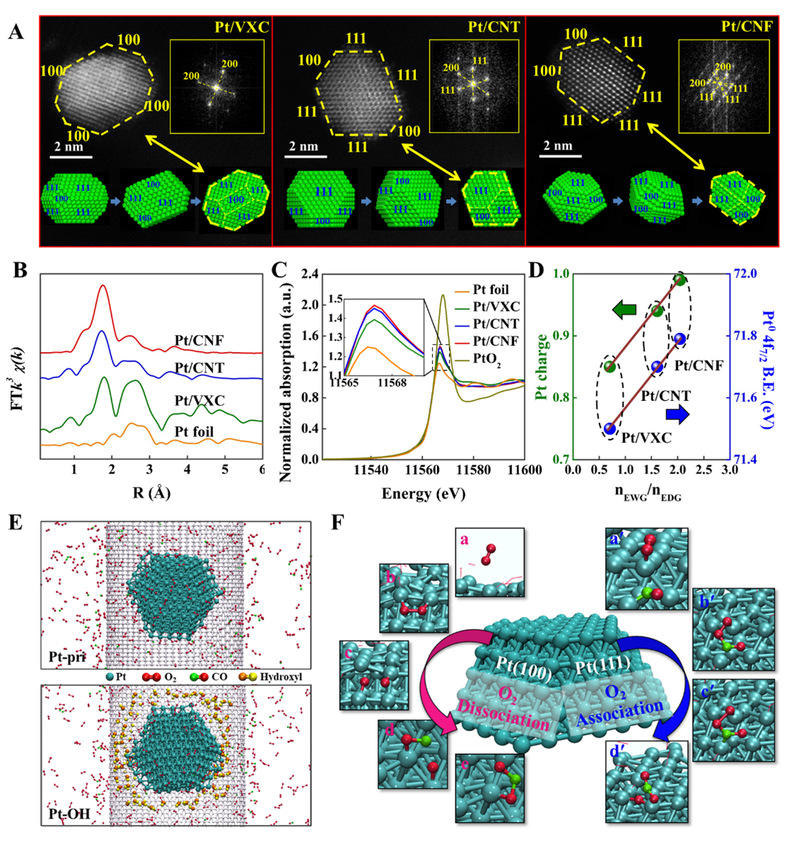
图1:炭载Pt催化剂的几何与电子结构表征及其分子动力学模拟
结果如图1所示,Pt催化剂表面CO氧化存在两种反应路径(O2缔合与O2解离)的竞争,且两种路径所占分率高度依赖于铂-炭载体界面间的电子分布,其中随着铂电子密度的降低,反应逐渐从O2解离路径过渡到O2缔合路径为主。与此同时,反应决速步骤(Rate-Determining Step, RDS)从O2*+*→2O*,过渡到O*+CO*→CO2*+*,最终演变为CO*+O2*→OOCO*+*。在此基础上提出了“自下而上”的研究策略,定量关联了金属-载体界面相互作用——反应机理——速率决速步的动力学与热力学信息——催化性能之间的关系(图2),系统揭示了金属-炭载体间的电子相互作用根源及其影响机制。
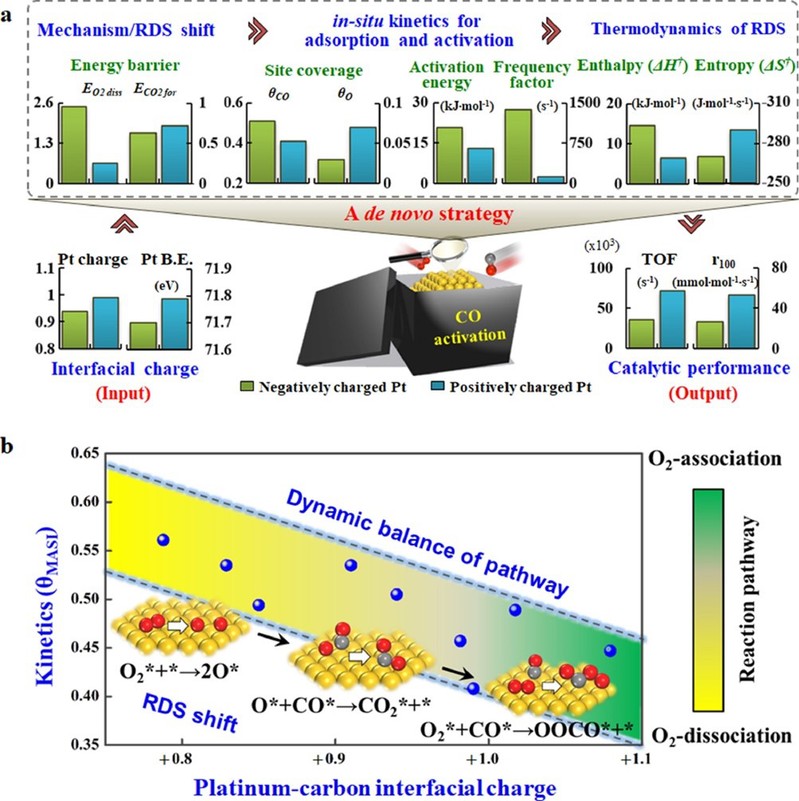
图2:基于“自下而上”策略的炭载Pt催化CO氧化动力学变化图
该研究得到了袁渭康院士、周兴贵教授与挪威科技大学De Chen院士等的悉心指导,以及我校特聘研究员练成与博士研究生刘长伟在理论计算模拟方面的帮助。此外,该研究得到了国家自然科学基金重大研究计划集成项目、国家自然科学基金优秀青年科学基金项目、上海教委科研创新计划自然科学重大项目等项目的经费支持。




